
This post originally appeared on Vector, Boston Children’s Hospital blog.
Screening a class of recently-developed drug compounds — so-called “CDK inhibitors” capable of blocking CDK7/12/13 proteins — against hundreds of different human cancer cell lines, researchers at Dana-Farber/Boston Children’s Cancer and Blood Disorders Center have found that CDK12 inhibitors pack a particularly lethal punch to Ewing sarcoma, a rare cancer typically affecting children and young adults.
“No one has previously considered CDK12 inhibition as a way to combat Ewing sarcoma,” says Kimberly Stegmaier, MD, senior author of the new Cancer Cell paper that describes the findings. In 2014, Nathaneal Gray, PhD, co-author on the new paper, and his team were the first to develop CDK inhibitors.
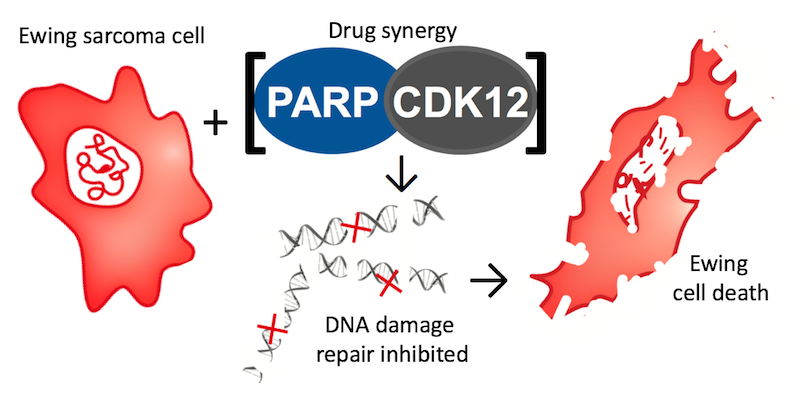
“Now, in mice, we’ve shown that Ewing sarcoma cells die if CDK12 is knocked out genetically or chemically inhibited,” Stegmaier says. What’s more, her team has discovered that CDK12 inhibition can be combined with another drug, called a PARP inhibitor, to double down on Ewing sarcoma cells.
The revelation that CDK12 inhibition can kill Ewing sarcoma cells brings a surge of hope to the field of pediatric oncology, which has long been challenged to find new drugs against childhood cancers.
“Pediatric cancers often involve abnormalities in genes that encode for transcription factors, shapeshifting proteins that bind to DNA sequences to activate or repress gene expression,” says Stegmaier, who co-directs the pediatric hematologic malignancy program at Dana-Farber/Boston Children’s and is a member of the Broad Institute’s Cancer Program. “Due to their disordered physical structure, transcription factors have largely eluded drug discovery efforts.”
A Molecular View of Ewing Sarcoma
In Ewing sarcoma, the second-most common bone cancer in children and adolescents, an error involving two genes gives rise to an abnormal fusion transcription factor called EWS/FLI. Once produced, EWS/FLI wreaks havoc by tripping the switch of normally quiet regions of DNA, turning on genes that are supposed to stay off, while also turning off genes that should be active.
“In Ewing sarcoma and other cancers involving transcription factor fusion ‘onco-proteins’ like EWS/FLI, these mutated transcription factors drive abnormal cell behavior and tumor growth,” Stegmaier explains. “EWS/FLI has so far not been able to be targeted by conventional drug chemistry.”
Cancer researchers call EWS/FLI a “pioneer factor” for its ability to turn on genes that wouldn’t be expressed otherwise.
But now, Stegmaier and her team may have unleashed a workaround to disarming EWS/FLI-expressing tumor cells via CDK12, a readily-targetable enzyme. After their wide-ranging drug screen revealed a potential link between CDK12 inhibition and Ewing sarcoma, the team sought to understand why.
“CDK12 is known to be important for gene regulation, so we wondered why the EWS/FLI fusion protein might engender sensitivity to CDK12 inhibitors,” Stegmaier says.
Getting To The Root of CDK12’s Role
In a mouse model, the team showed that CDK12 inhibitors greatly slow down tumor growth and extend the survival rates of mice with Ewing sarcoma tumors.
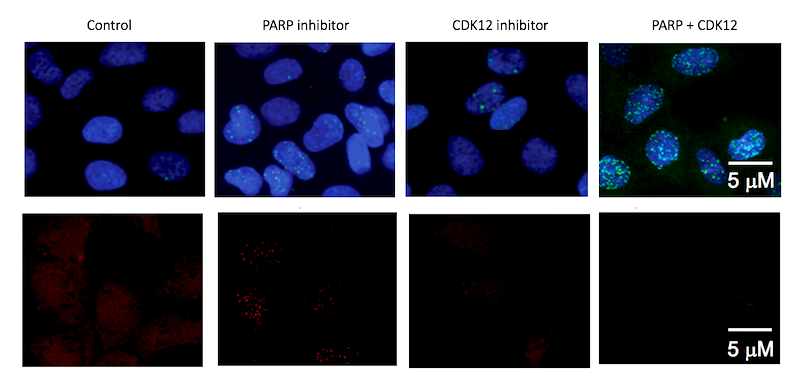
Searching through the scientific literature, the team found that other cancer researchers had documented a link between CDK12 and ovarian cancer. Ovarian tumors with a genetic mutation inactivating the CDK12 protein are exquisitely sensitive to FDA-approved drugs called PARP inhibitors, which block the activity of PARP proteins involved in DNA damage repair and other cellular processes.
Stegmaier’s project team, led by postdoctoral fellow Amanda Balboni Iniguez, PhD, hypothesized that inhibiting CDK12 in Ewing sarcoma might have a double-whammy clinical benefit: slowing down the growth of Ewing cells and also making them lethally sensitive to PARP inhibitors.
A New Drug Cocktail for Ewing Sarcoma?
“When we combined CDK12 and PARP inhibitors in Ewing sarcoma cells in culture dishes and in mouse models of Ewing sarcoma, we saw a very dramatic effect; some individuals were entirely cured of the disease,” Stegmaier says. “We have discovered that CDK12 inhibitors repress genes important to the regulation of DNA damage in Ewing sarcoma cells, and therefore these EWS/FLI positive Ewing sarcoma cells are very sensitive to PARP inhibitors.”
What’s more, the CDK12 and PARP inhibitor drug combination had no toxic effect on the bone marrow of the mice. In contrast, PARP inhibitors given in combination with chemotherapy have already shown to create a high amount of bone marrow toxicity in humans.
Given these experimental results, Stegmaier is excited about the future potential for testing the combination of CDK12 and PARP inhibitors in clinical trials.
“PARP inhibition is already FDA-approved for use in certain cancers, and inhibitors of CDK12 and other CDK proteins are in early-phase clinical testing,” Stegmaier says. “If they are proven to be safe in adults, I hope we can extend testing to children with Ewing sarcoma.”
In addition to Stegmaier, Gray and Balboni, the paper’s other authors are: Björn Stolte, Emily Jue Wang, Amy Saur Conway, Gabriela Alexe, Neekesh V. Dharia, Nicholas Kwiatkowski, Tinghu Zhang, Brian J. Abraham, Jaume Mora, Peter Kalev, Alan Leggett, Dipanjan Chowdhury, Cyril H. Benes and Richard A. Young.
This work was supported by the Brian MacIssac Sarcoma Foundation, the Ty Louis Campbell Foundation, the National Cancer Institute (1R35 CA210030), the St. Baldrick’s Foundation, National Institutes of Health (HG002668), the Koch Institute and Dana– Farber/Harvard Cancer Center Bridge Grant and the Damon Runyon Foundation Fellow.