
This post was originally published on Vector, Boston Children Hospital’s science and clinical innovation blog.
In 1938, Louis K. Diamond, MD, and Kenneth Blackfan, MD, at Boston Children’s Hospital described a severe congenital anemia that they termed “hypoplastic” (literally, “underdeveloped”) because of the bone marrow’s inability to produce mature, functioning red blood cells. Eighty years later, the multiple genetic origins of this highly rare disease, now known as Diamond-Blackfan anemia, or DBA, are finally coming into view.
The largest study to date, published recently in the American Journal of Human Genetics, raises as many questions as it answers. But in the meantime, it provides a genetic explanation for nearly 80 percent of patients.

One of them, 5-year-old Jack Farwell, recently came to the Dana-Farber/Boston Children’s Cancer and Blood Disorders Center for a blood transfusion. As always before a transfusion, he was pale and tired, but also cheerful and talkative, showing off Pinchy Mars and Red Claw, his stuffed crabs.
Jack was born prematurely at just 27 weeks. Doctors in the NICU noticed his hemoglobin was low, meaning he had very few red blood cells. Despite having a blood transfusion, Jack’s hemoglobin was down again within a couple of weeks.
Several transfusions later, Jack had a bone marrow biopsy. It indicated that he had few red-cell precursors, the hallmark of DBA, and would need monthly transfusions, indefinitely, to replenish his red cells. Since the transfusions eventually cause iron overload, his doctors prescribed iron chelators. They also installed a port to make needle insertion easier.
“They were having to bring in an IV specialist, he was so little and his veins were so picked over,” says Lee Farwell, Jack’s mother. “The port did help.”
Vijay Sankaran, MD, PhD, in the hematology program of the Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, checks Jack’s vitals. They discuss another of Jack’s current interests: bones.
“Do you know what happens inside the bones?” Sankaran asks. “Blood is produced.”
Except that Jack’s bones don’t produce red blood cells. Why?
Investigating DBA genetics
Twenty years ago, Hanna Gazda, MD, PhD, started on the path of looking comprehensively at DBA’s genetics. Her goal was to better understand its causes, so that better treatments could be developed. Other than lifelong transfusions, the main treatment is steroids, but about half of patients, including Jack, don’t benefit, and steroids can cause unpleasant side effects.
At the time, just one DBA-related gene was known. Gazda began gathering patients with the condition in her native Poland, while at the Medical University of Warsaw. She continued after establishing her own lab to Boston Children’s Hospital in 2010, part of the Division of Genetics and Genomics and the Manton Center for Orphan Disease Research.
With support from her mentors Colin Sieff, MB, BCh, of the Dana-Farber/Boston Children’s hematology program, and Alan Beggs, PhD, director of the Manton Center, Gazda identified 10 additional DBA genes. That gave a clue: Like the first identified gene, all the genes code for proteins that make up the ribosomes, the cellular organelles that themselves build proteins.

“We sequenced many ribosomal protein genes and looked for large chromosomal deletions, and were able to find mutations for about 60 percent of patients,” says Gazda. “We confirmed that it is a ribosomal disease.”
But a mystery remained. Ribosomes are in virtually all our cells, and they make many different proteins. What do they have to do with red blood cells specifically? Why aren’t other kinds of cells affected in DBA?
Another mystery: Why do steroids like cortisone often work in DBA? They were first tried in the 1960s, a last-ditch measure at a time when steroids were being tried for many diseases.
“Suddenly these patients could make red cells,” recounts David Nathan, MD, president emeritus of Dana-Farber Cancer Institute, who has devoted much of his career to DBA. “That was startling. The question was, why?”
Connecting the dots with GATA1
Gazda met Sankaran soon after arriving at Boston Children’s. He suggested teaming up, given his group’s interest in utilizing emerging genomic approaches such as whole-exome sequencing. By this time, with the help of many collaborators, Gazda had amassed samples from 472 families from Europe, the U.S. and elsewhere. Together, they’re thought to represent a quarter to a third of all people with DBA.
The added genetic power enabled new insights. In 2012, Gazda and Sankaran found the first non-ribosomal DBA mutation, affecting a gene called GATA1. This piqued their interest, since Stuart Orkin, MD, at Boston Children’s had identified GATA1 decades ago as being essential for precursor cells to differentiate into red blood cells, following Nathan’s 1978 discovery that DBA is caused by these precursors being trapped in an early stage of development. Yet no other GATA1 mutations had been discovered in DBA.
Sankaran subsequently led studies, published in Cell and Nature Medicine, that helped tie the GATA1 and ribosome findings together. Together they showed, for the first time, that DBA mutations reduce the actual numbers of ribosomes in blood precursor cells. Without enough ribosomes, the precursors can’t produce enough GATA1, so mature red cells never form. Other blood cells — like platelets, T cells and B cells — can still develop since they’re not dependent on GATA1.
“Sankaran had a brilliant idea – that certain messages, complicated messages, don’t get translated when you don’t have enough ribosomes,” says Nathan. “He showed that the messenger RNA for GATA1 is there, but it can’t get translated, while other messages can. That created completely new thinking about biology, protein synthesis in general, and the role of the ribosomes in clinical medicine.”

Diamond-Blackfan anemia: A 30,000 foot view
In the new paper, Gazda, Sankaran and their colleagues provide a comprehensive snapshot of DBA. Using whole exome sequencing combined with a slate of other methods, they found a genetic diagnosis for 78 percent of their 472 patients. They further confirmed the role of ribosomal protein genes, finding mutations in the 19 known genes (many discovered by Gazda), plus seven genes not previously recognized. The study also revealed that some patients with a DBA diagnosis actually have a different disease.
With such a large sample, Gazda and Sankaran can now begin grouping the different mutations and matching them with symptoms. Many patients with DBA have other medical issues, such as heart problems, physical abnormalities like cleft lip/palate and an increased risk for certain cancers.
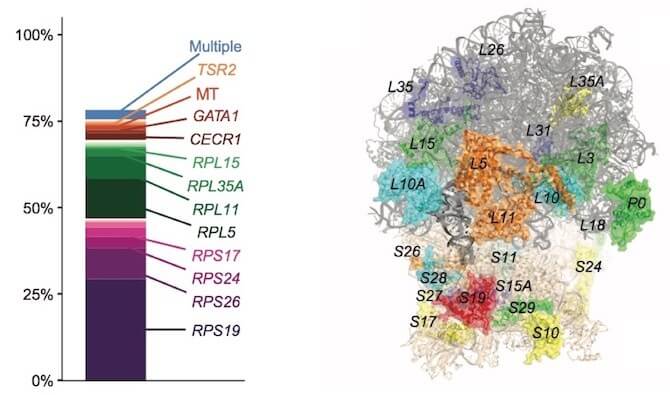
At left, the percentage of presumed causal mutations in each gene, totaling 78 percent for the cohort as a whole. At right, the structure of a ribosome highlighting the 19 known and 7 new genes that are mutated in its component proteins. The color coding reflects the number of known mutations: blue (none found in this study, but reported in other studies), cyan (1 mutation), green (2–3 mutations), yellow (7–15), orange (31–54), and red (more than 100). (Not seen from this angle: RPS7, RPL27, RPL34, and RPL19). (ILLUSTRATIONS ADAPTED FROM AJHG 2018: DOI 10.1016/j.ajhg.2018.10.027)
Jack’s mutation, which is in one of the 19 ribosomal proteins, took about a year to find. His parents were relieved to learn that it was a spontaneous mutation, so any of their future children would not be at risk for inheriting it. The mutation also impairs Jack’s ability to produce antibodies, leading to multiple severe infections. He now receives regular antibody infusions at home.
“This goes along with his type of mutation,” says Sankaran. “About 40 patients with similar mutations have been studied and many of them require antibody replacement too. We don’t understand exactly why. But we now know better which mutations might cause which features of DBA.”
The study also helps understand the “penetrance” of DBA mutations: Not everyone found to have a known mutation actually has a clinical illness.
“There’s very large clinical variability,” says Gazda. “In one family, there can be one person with severe anemia and another with mild anemia. This could lead to a new avenue of research: some patients may have modifier genes that affect disease severity.”
Translating mutations to a treatment?
Despite the size and power of Gazda’s cohort, we’re still largely at the beginning. The 26 known ribosomal protein genes represent only about a third of the genes involved in forming ribosomes. And even though the study found mutations in GATA1 and a handful of other genes, 20 percent of patients still have no known genetic cause.
One patient originally thought to have DBA proved to have an even rarer anemia caused by a mutation in his erythropoietin (EPO) gene. He and his younger sister were successfully treated with recombinant EPO, which helps stimulate red-cell production.
Finally, to develop better treatments — gene therapy, a drug treatment or something else to stimulate red-cell production — researchers still need to know more about DBA’s biology.
“We can’t develop 26 different gene therapy vectors,” say Sankaran. “We need a strategy that’s going to target all of these patients. With the discovery of GATA1 being a common downstream mechanism, we could look into upregulating GATA1. There’s some evidence that this rescues red blood cell production, at least in vitro.”
Gene therapy pioneer David Williams, MD, PhD, president of Dana-Farber/Boston Children’s, agrees.
“This is a classic example of how basic research can lead to innovations in treatment,” says Williams, who is also Boston Children’s chief scientific officer and senior VP for research. “At Boston Children’s, we excel at this, and with the new knowledge of the importance of GATA1 as a downstream integrator of DBA’s pathophysiology, we can now approach treating this disease in an effective and efficient manner.”
Gazda and Sankaran hope to gain further biological insights through whole-genome sequencing. Unlike whole-exome sequencing, this technique sequences the “non-coding” portions of DNA, which often contain regulators of genes. “It definitely can bring more insights,” Gazda says.
Delving into steroids
Meanwhile, Nathan still wants to know why steroids often work in DBA. Hojun Li, MD, PhD, an attending physician at Dana-Farber/Boston Children’s and a fellow in the lab of Harvey Lodish, PhD, at MIT, is among those exploring this question.
“We really never knew the biochemical basis of steroids’ action,” says Nathan. “Somehow they must improve the translation of GATA1. Steroids trigger the activity of a lot of genes, so there must be a steroid-sensitive gene that helps translate messages from GATA1 to stimulate the growth of red blood cell progenitors.”
Ultimately, if researchers find such a gene, we can then try find a drug that acts like cortisone, but doesn’t have the side effects, Nathan says.
“Our study starts to address some questions about Diamond-Blackfan anemia, but it also raises just as many questions,” says Sankaran. “Research is never an endpoint. It’s a constant yearning to figure out more.”
Jacob Ulirsch, of the Manton Center and Dana-Farber/Boston Children’s hematology program, was first author on the current study. This work was supported by the National Institutes of Health, the DBA Foundation, the New York Stem Cell Foundation, The French National Research Agency (ANR) and the European Research Arena (ERA-NET). See the paper for a full list of authors and grants.